Familial
Hypercholesterolemia



Familial hypercholesterolemia (FH) is a genetic disorder of the lipoprotein metabolism, which
is characterized by significantly
elevated low-density lipoprotein-cholesterol
LDL-C
![]() . 1,2 FH includes
. 1,2 FH includes
Familial: The underlying cause is a genetic defect that is passed down through families
Hypercholesterolemia: High LDL-cholesterol
In patients with FH, chronic exposure to high circulating cholesterol levels
is a risk factor that leads to1,2

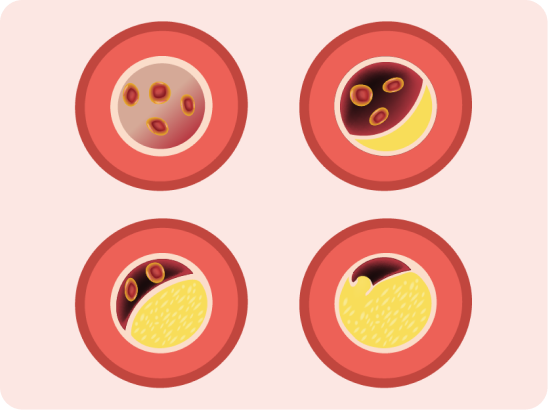
Atherosclerotic plaque
![]() deposition in the coronary arteries and proximal aorta
deposition in the coronary arteries and proximal aorta
Increased risk of (premature) atherosclerotic cardiovascular disease (ASCVD) (e.g. angina, myocardial infarction, stroke)
High mortality rates
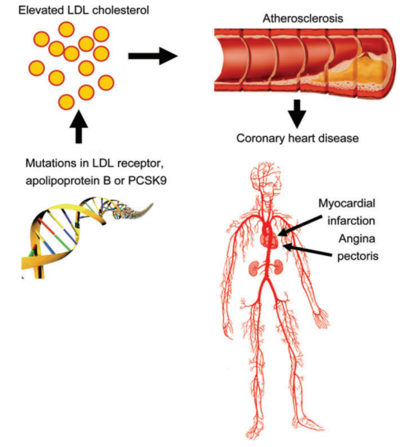
Adapted from Nordestgaard BG, et al. Eur Heart J. 2013
FH is inherited as a monogenic autosomal dominant trait (Read more about genetics of FH here). Patients with FH can be categorized as having
Homozygous familial hypercholesterolemia (HeFH)

Homozygous familial hypercholesterolemia (HoFH)

An estimated
30 million people worldwide could be affected by FH
but prevalence estimates between studies vary substantially.3
Global FH prevalence:*
4 in 1000 people3
*pooled estimate of 0.4%
Asia FH prevalence:*
5 in 1000 people3
*pooled estimate of 0.46%
Patients with HoFH or compound HeFH present with distinctive and severe clinical manifestations very early in life (< 10 years old). On the other hand, patients with HeFH are, by and large, asymptomatic in childhood and adolescence and typically diagnosed by screening methods.7
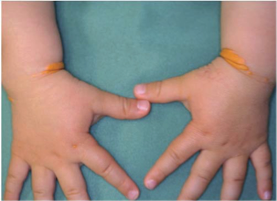
Xanthomas are the most common skin lesions in FH. They are composed of
monocyte
![]() -derived
foam cells (accumulation of
lipids inside the cells) and connective tissue.
-derived
foam cells (accumulation of
lipids inside the cells) and connective tissue.
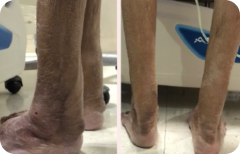
Achilles tendon xanthoma
"Skin-colored subcutaneous nodule, smooth and firm, attached on Achilles tendon"

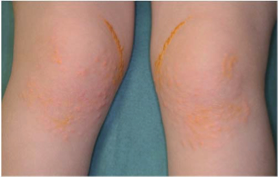
Tuberous xanthoma
"flat, or elevated and rounded, yellowish nodules on the skin over joints,
especially on elbows and knees"
Cutaneous xanthoma8 in a 3-year-old patient at flexures of the wrist and in other regions with mechanical stress.
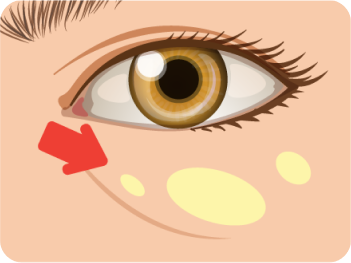
Xanthelasma are the most common type of cutaneous xanthomas. They are sharply
demarcated, yellowish, flat or minimally elevated, soft/semisolid plaques. It is
distributed symmetrically, occurs most commonly near the inner
canthus
![]() of the eyelid, more often
on the upper rather than the lower lid.
of the eyelid, more often
on the upper rather than the lower lid.
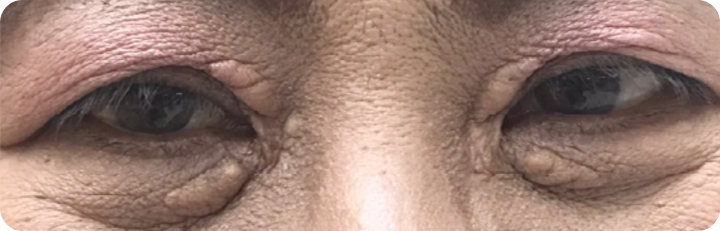
*courtesy of images from patients at King Chulalongkorn Memorial Hospital
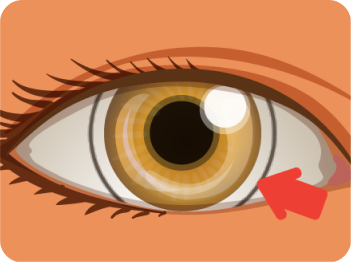
Corneal Arcus
![]() is described as a gray-white-yellowish opacity, 1–1.5 mm thick,
located at the periphery of the cornea. It is formed by the
deposition of lipids in the stroma of cornea.
is described as a gray-white-yellowish opacity, 1–1.5 mm thick,
located at the periphery of the cornea. It is formed by the
deposition of lipids in the stroma of cornea.
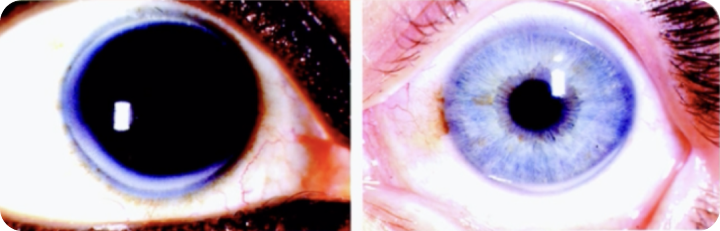
*courtesy of images from patients at King Chulalongkorn Memorial Hospital
including multiple vascular beds, including coronary, cerebral, and peripheral vascular system.

including
aortic stenosis
![]() due to cholesterol and inflammatory cell infiltration.
due to cholesterol and inflammatory cell infiltration.
FH is inherited as a monogenic autosomal codominant trait, so patients with FH can be categorized as HeFH or HoFH. Mutations in key proteins involved in LDL metabolism are the underlying cause of FH, and lead to reduced uptake of LDL by hepatocytes and thus elevated serum LDL-C levels.1,2,4

Heterozygous familial
hypercholeste
rolemia
(HeFH)

Homozygous familial
hypercholeste
rolemia
(HoFH)

(Heterozygous or Homozygous)
mutations in genes encoding key
proteins involved in LDL uptake
Elevated
plasma LDL-C
Defective LDL uptake in hepatocytes
Key proteins involved in LDL uptake9,11
LDL in blood stream is taken up by hepatic and extra-hepatic tissues through LDL-receptors locating on the cell membrane. There are four proteins involved in this process.
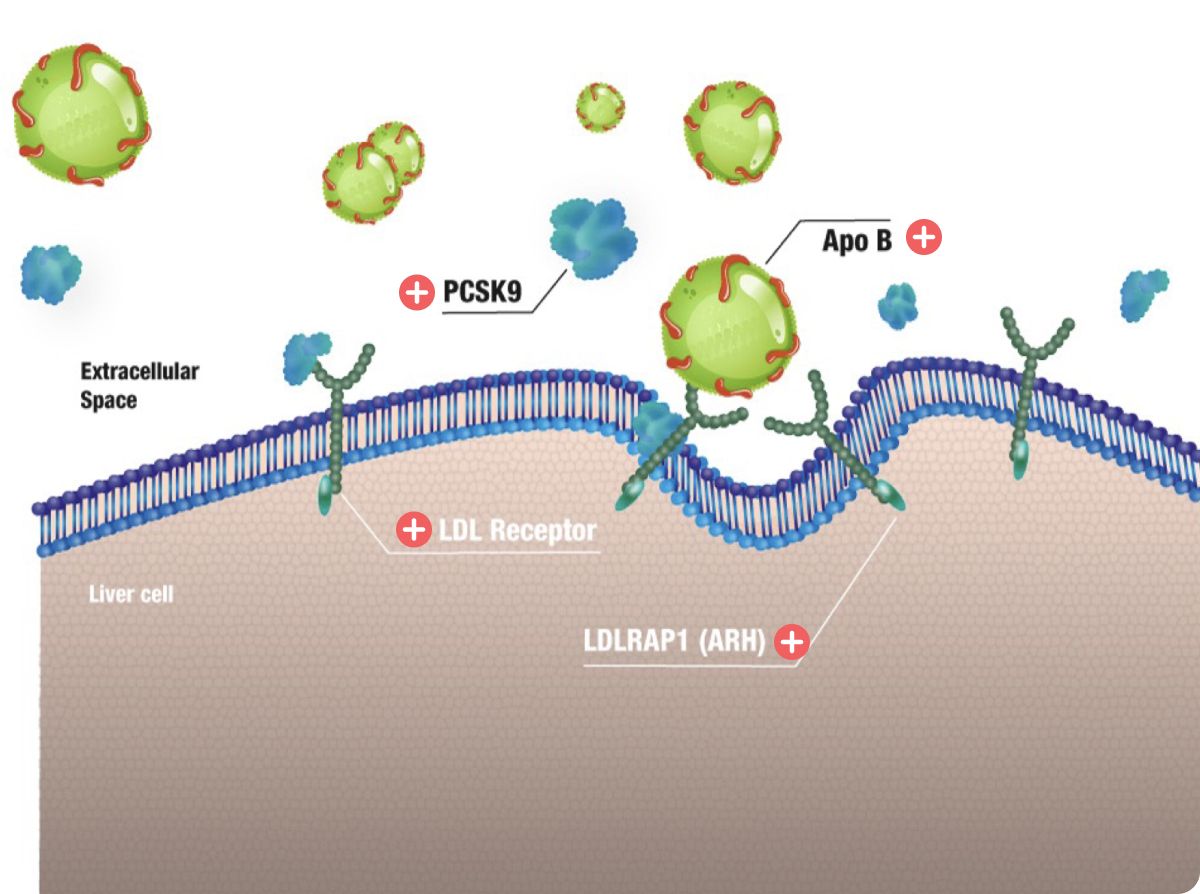
LDLR, low-density lipoprotein receptor; LDLRAP1, LDLR adaptor protein 1; PCSK9, proprotein convertase subtilisin/kexin type 9
| Gene | Mechanism of gene mutation | Prevalence |
|---|---|---|
| LDLR | LDLR is absent or has decreased capacity to clear LDL from the circulation | 85–90% (most common) |
| APOB | Mutations impair binding of LDL to the LDLR, reducing LDL uptake | 5–10% |
| PCSK9 | Gain-of-function mutations increase PCSK9 activity, leading to increased LDLR degradation and decreased surface expression of LDLR, thus reducing uptake of LDL | Rare |
| LDLRAP1 | Loss-of-function mutations in the protein required for clathrin-mediated internalization reduce the uptake of the LDLR–LDL complex | Rare |
FH is associated with a significantly increased CVD risk because elevated low-density lipoprotein cholesterol (LDL-C) in FH leads to atherosclerotic plaque deposition in the coronary arteries and proximal aorta at an early age and increases the risk of premature cardiovascular events.1

Elevated plasma LDL-C
Cholesterol retention in the arterial
wall and foam cell formation within
the intima of arteries lead to
occlusive atherosclerosis
Patients with FH are at significantly
increased risk of CHD and MI
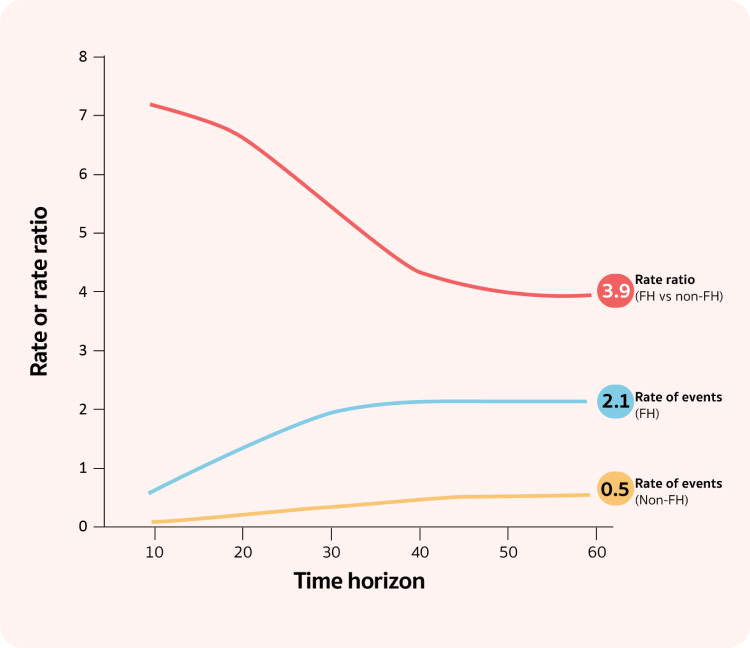
Lifetime CVD Event Rates in FH
is 4 times higher than
Non-FH Patients14

Untreated patients with HoFH are likely to develop
CHD as teenagers and die from acute MI as early as 4
and before reaching 20 years of age15
Adults with the FH phenotype, defined as LDL-C
≥ 190 mg/dL, were associated with ~5-fold increase
in long-term CHD and ASCVD risk compared with
those with LDL-C < 130 mg/dL16